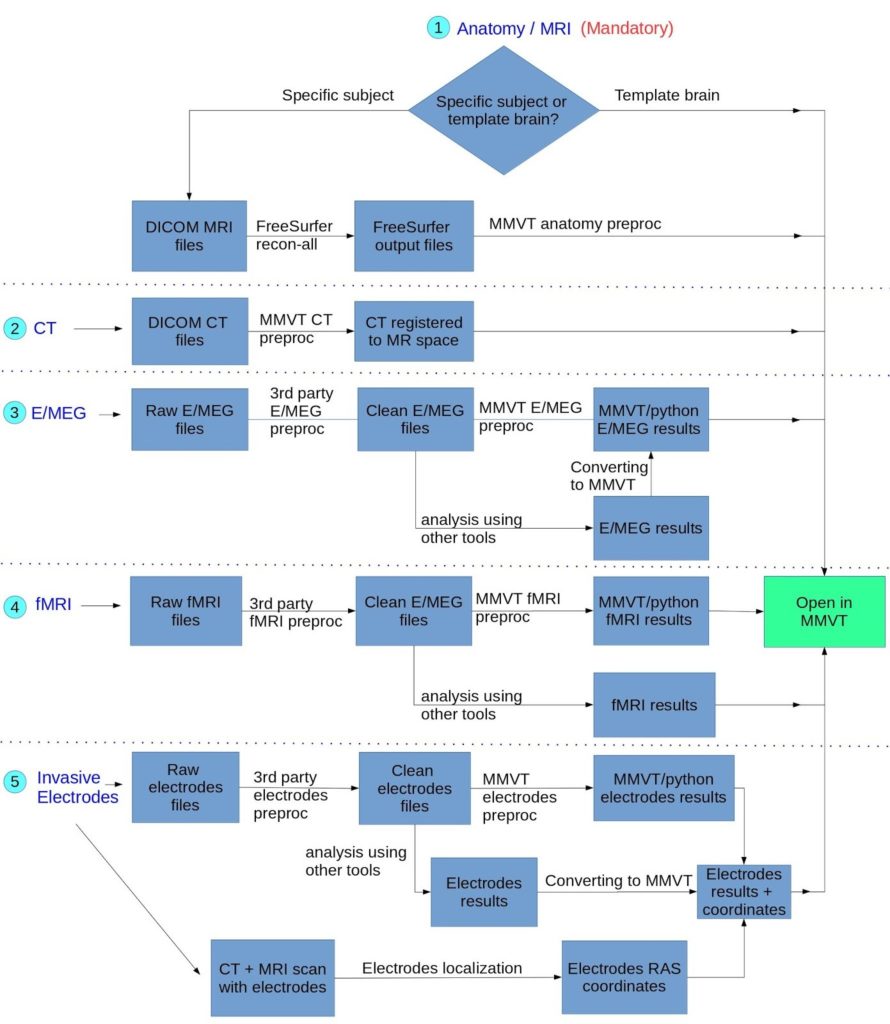
If you wish to load FsFast results, the script will try to read the FsFast directories tree:
bold/{contrast_name}.sm05.{hemi}/{contrast}/sig.nii.gz
Also, the script assumes those directories are placed in the fMRI link directory, so the full directory tree should look as following:
links/fMRI/{task}/{subject-name}/bold/{contrast-name}.sm05.{hemi}/{contrast}/sig.nii.gz
You need to specify the task and the contrast name. You can also specify a contrast, or leave it empty to get all the contrasts:
python -m src.preproc.fMRI -s subject-name -a atlas-name --contrast_name contrast-name -t task-name -f fmri_pipeline
The atlas-name is needed for the clustering step. The algorithm will find cortical clusters and calculate their intersections with the labels, according to the selected atlas. In case the recon-all files are in a remote directory, like in the [anatomy preprocessing] (https://github.com/pelednoam/mmvt/wiki/Anatomy-preprocessing), you can set the remote_subject_dir:
python -m src.preproc.fMRI -s subject-name -a atlas-name --contrast_name contrast-name -t task-name -f fmri_pipeline --remote_subject_dir full-path-to-subject-recon-all-files
By default, the threshold for calculating the clusters is set to 2. If you wish to change it, you should use the threshold flag:
python -m src.preproc.fMRI -s subject-name -a atlas-name --contrast_name contrast-name -t task-name -f fmri_pipeline --threshold threshold-value
By default, the colors will be calculated symmetrically, from -abs(max(data)) to abs(max(data)). That makes sense if you loads fMRI contrast map. In case you want the color to be calculated from min(data) to max(data), you should use the symetric_colors flag:
python -m src.preproc.fMRI -s subject-name -a atlas-name --contrast_name contrast-name -t task-name -f fmri_pipeline --symetric_colors 0
If you wish to rerun the script for the same subject, and overwrite the previous results, you should use the overwrite_surf_data flag:
python -m src.preproc.fMRI -s subject-name -a atlas-name --contrast_name contrast-name -t task-name -f fmri_pipeline --overwrite_surf_data 1
If you wish to run the script on more than one subject, you should write the subjects names, separated with a comma, without spaces. If you also need to specify the remote directory, you should add {subject}:
python -m src.preproc.fMRI -s subject1-name,subject2-name,subject3-name -a atlas-name --contrast_name contrast-name -t task-name -f fmri_pipeline --remote_subject_dir full-path-to-subject-recon-all-files/{subject}"
Make sure you are using apostrophes in the remote folder name, the ‘{‘ character can be problematic.
In case you just want to load fMRI/PET volume file (nifty image), the preprocessing script will also project the volume data on the hemispheres:
python -m src.preproc.fMRI -s subject-name -a atlas-name
In this case, the volume file will be located in:
links/fMRI/subject-name/subject-name.{format}
Where format can be nii, nii.gz or mgz. In case you want to set the volume file name, you should use the volume_name flag:
python -m src.preproc.fMRI -s subject-name -a atlas-name --volume_name volume-name
Now the volume file will be located in
links/fMRI/subject-name/volume-name.{format}
In case the files are located under a task folder, like in the FsFast example, you should set the task flag:
python -m src.preproc.fMRI -s subject-name -a atlas-name --volume_name volume-name -t task-name
Now the volume file will be located in
links/fMRI/task-name/subject-name/volume-name.{format}
In case you wish to load PET data, you need to use the is_pet flag. Also, in PET raw data, you most probably don’t want the colors to be calculated symmetrically, and the threshold should be set to a meaningful value, possibly zero:
python -m src.preproc.fMRI -s subject-name -a atlas-name --volume_name volume-name -t task-name --threshold 0 --symetric_colors 0 --is_pet 1
First, you should call the clean_4d_data function:
python -m src.preproc.fMRI -s subject-name -f clean_4d_data --fmri_file_template=file-template
Where file-template is the usually a wildchar expression for finding the raw file in the subject’s fMRI folder. You can also create a link to the original file. For example, if your file’s name is f.nii, use can set the –fmri_file_template to be “f.*”.
This function is basically a wrapper for the following freesurfer calls: preproc-sess, tkregister-sess, fcseed-config, fcseed-sess and selxavg3-sess. Those calls require a functional subdirectory, which as default is ‘rest’, but you can change it using the –fsd flag. Also, the default values for fwhm, lfp and nskip are 6, 0.08 and 4 respectivly, and you can change them using the flags –fwhm, –lfp and –nskip.
Usually, these freesurfer functions are morphing the data into the template brain. For doing so, you need to set the –template_brain flag to a template brain, like fsaverage, or fsaverage5. If you are not setting this flag, the data will remain in the subject’s MRI space. This option is preferable if later you want to import the data into the subject’s MMVT file. The function’s output is two files, one for each hemisphere: ‘{}.sm{}.{}.{}.mgz’.format(fsd, int(fwhm), trg_subject, hemi). If you’ve picked the default values, didn’t morph the data to a template brain, and set fsd to rest for example (if the data is resting state fMRI), the files will be named rest.sm6.subject-name.rh.mgz and rest.sm6.subject-name.lh.mgz
Next, if you wish to extract the time series from the cortical labels, you should call the analyze_4d_data function:
python -m src.preproc.fMRI -s subject-name -a atlas-name -f analyze_4d_data --fmri_file_template "*.{hemi}.{format}"
You can set the fmri_file_template to be more specific like ‘restsm6{hemi}.mgz’ for example. The function will calculate the mean of each label for each time point. If you don’t want to calculate the mean for all the labels, you can set the –excluded_labels flag. The default value is ‘corpuscallosum,unknown’. If you wish to calculate a different measure than the mean, you can set the –labels_extract_mode flag, where the possible values are mean,pca and cv (coefficient of variation). You can use more than one (mean,cv for example).
If you have the recon-all files of your subject, you just need to use the same approach like in fMRI/PET raw data. In case you don’t have the subject’s MRI, you’ll need to morph the result in SPM to the SPM template brain. Then, use colin27 as your subject-name when calling to the fMRI preprocessing (you’ll need to download colin27 first from here:
python -m src.preproc.fMRI -s colin27 -a atlas-name
If you wish to load group averaging/comparison results, you first need to convert the SPM results to FreeSurfer format. In the SPM directory, you’ll find spmT_0001.img and spmT_0001.hdr. In the command prompt, you’ll need to run the following command (after sourcing FreeSurfer):
mri_convert spmT_0001.img results-name.nii
Where results-name can be the subject’s name in case of subject vs group comparison, or any other name you find suitable. After that, copy the nii file to colin27’s fMRI results folder, which can be (as described above) in links/fMRI/colin27 or in links/fMRI/task-name/colin27 if you specify the task (using the -t flag). The file should be copied to the colin27 folder because, for group averaging/comparison, SPM uses its template brain, which is colin27. After that, you can load this file as described in the loading fMRI/PET raw data section.
This section will be described more thoroughly in the MMVT addon documentation. For now, if you wish to render your data from the command prompt, you can run the following script:
python -m src.mmvt_addon.scripts.render_fmri_clusters -s subject_name -a atlas_name
The end result of this script is a split brain figure, with lateral and medial views:

This script will render the clusters for all the subject’s contrast you’ve calculated. If you are using colin27, that will render all the clusters for all the results loaded to colin27. For example, if you used SPM to calculate the difference between several subjects and a control group, the contrasts will be the subjects names, and the script will render the results of all the comparisons. Also, by default, it’ll render both the pial and half inflated brain and both in white and black backgrounds. For more details, take a look at the script’s documentation. If you are using a specific subject (not colin27), you’ll need to create the medial and lateral views manually, as described in the rendering panel documentation.
Given the low time resolution, only resting state data is supported for functional connectivity. To analyze the resting state fMRI data, you need to call the fMRI preprocessing as the following:
python -m src.preproc.fMRI -s subject-name -a atlas-name -f analyze_resting_state --fmri_file_template fmri-file-template
This call will load an fMRI file according to the given a template for each hemisphere, and calculate a time series for each label according to the given atlas (atlas-name). This function assumes that the data was already projected on the cortical surface. If this isn’t the case, you should first project it, by calling the fMRI preprocessing as described here. You just need to add the -f project_volume_to_surface flag to the call, to run only the projection function. For example, let’s say you’ve already processed your fMRI data with FsFast, and as result, you got two files, subject-name.siemens.sm6.fsaverage.rh.nii.gz and subjext-name.siemens.sm6.fsaverage.rh.nii.gz where subject-name is the name of your subject. First, you need to copy those files to the links/fMRI/subject-name/ folder. The fmri-file-template is {subject}.siemens.sm6.{morph_to_subject}.{hemi}.{format}. The code will put subject-name in {subject} and rh and lh in {hemi}. The default value for the format is ‘nii.gz’. In case you want to set a different format, you should use the –input_format flag. The {morph_to_subject} is used in case the data was projected to a template brain, fsaverage, like in the current example. In such a case, you’ll need to use the flag –morph_labels_to_subject. You can also use wildcards in the fmri-file-template. For example: “{subject}*{morph_to_subject}.{hemi}*.{format}”. That will work only if there is only one file matching this template. If your files are named ‘subject-name.siemens.sm6.fsaverage.rh.b0dc.nii.gz’ for example, this template will catch it. The default measure to calculate a time series for each label is just taking the mean of all the label’s vertices. The other option is ‘PCA’, where the first principal component is taken. To change this option you’ll need to use the –labels_extract_mode flag. The default atlas-name is aparc.DKTatlas40, as explained here. To summarize, in the current example the call is the following:
python -m src.preproc.fMRI -s subject-name -a atlas-name -f analyze_resting_state --fmri_file_template "{subject}\*{morph_to_subject}.{hemi}\*.{format}" --morph_labels_to_subject fsaverage
The resting state analysis function will create two files, one for each hemisphere: labels_data_{atlas-name}{labels-extract-mode}{hemi}.npz. The files are located in the links/mmvt_blend/subject-name/fmri folder. These npz files include two variables: data (labels_num x T), and names, the labels names.
If you wish to call this function in code, look at the function analyze_resting_state in src.preproc.examples.fMRI, or call it directly:
python -m src.preproc.examples.fMRI -s subject-name -a atlas-name -f analyze_resting_state
Remember that if you are using the aparc.DKTatlas40 atlas, you don’t need to use the -a flag.
If you don’t know the TR, you can user the FreeSurfer mri_info utility, or call this function:
python -m src.preproc.fMRI -s subject-name -f get_tr --fmri_fname fmri_fname
Where fmri_fname is your fMRI file’s name, like subject-name.fsaverage.rh.nii.gz for example.The only two supported file types are nii.gz and mgz. If fmri-fname type is nii.gz, the code will convert it to mgz.
The fMRI preprocessing creates two files, one for each hemisphere: labels_data_(atlas_name}{labels_extract_mode}{hemi}.npz. The files are located in the links/mmvt_blend/subject-name/fmri folder. These npz files include two variables: data (labels_num x T), and names, the labels names. To load them and run a functional connectivity analysis, you should call the connectivity preprocessing module as the following:
python -m src.preproc.connectivity -s subject-name -a atlas-name -f calc_lables_connectivity --connectivity_modality fmri --connectivity_method connectivity-method --windows_length windows-length --windows_shift windows-shift
Where the connectivity-method is the method you want to use to calculate the functional connectivity. The methods that are supported are Pearson correlation (corr) and Phase Lag Index (pli).The calculation will be done using sliding windows, according to the window-length (the length of the windows) and window-shift (the shift in each slide). You can also calculate the coefficient variation of the values by concatenating ‘cv’ (using a comma) to the connectivity-method. The unit of the length and shift of the windows is the fMRI TR. For example, if your fMRI TR is 3s, windows-length=20 and windows-shift=3, then the length of the windows will be 60s, and the windows shift 9s.
The end result is the following files in the links/mmvt_blend/subject-name/connectivity:
- fmri_connectivity-method.npy: The numpy matrix, NxNxW, where N is the number of the cortical labels and W is the number of windows.
- fmri_connectivity-method.npz: The connectivity properties for visualization.
- fmri_vertices.pkl: The vertices information for visualization.
In case you added cv to the connectivity-method, these files will also be created:
- fmri_connectivity-method_cv.npy: The coefficient variation of the fmri_connectivity-method matrix.
- fmri_connectivity-method_cv.npz: The coefficient variation connectivity properties for visualization.
- fmri_connectivity-method_cv_mean.npy: The mean of the fmri_connectivity-method_cv matrix.
- fmri_connectivity-method_cv_mean.csv: A cortical labels coloring file, which is saved in the ../coloring folder.
For example, if you wish to:
- Calculate the correlation between the fMRI BOLD signal of the cortical labels time course.
- Where the labels are parcellated according to the Lausanne125 atlas
- Calculate also the coefficient of variation for each label
- Use windows length of one minute and shift of 9s, where the TR is 3s
python -m src.preproc.fMRI -s subject-name -a laus125 -f calc_lables_connectivity --connectivity_modality fmri connectivity_method=corr,cv --windows_length 20 --windows_shift 3
If you wish to call this script from code, take a look at the example in src.preproc.examples.connectivity.calc_fmri_connectivity:
from src.preproc import connectivity as con
args = con.read_cmd_args(dict(
subject=args.subject,
atlas='laus125',
function='calc_lables_connectivity',
connectivity_modality='fmri',
connectivity_method='corr,cv',
windows_length=20,
windows_shift=3
))
con.call_main(args)